- Seizures are defined as a paroxysmal occurrence of abnormal excessive and/or synchronous neuronal activity of the brain. This often, but not always, causes neurological symptoms.
- Seizures are not a disorder in and of themselves but are a symptom of various neurological or systemic etiologies.
- The first step in seizure classification is to identify whether seizures are focal or generalized in onset.
Focal onset seizures
- Previously called partial seizures, focal onset seizures can be subclassified as focal aware or focal with impaired awareness.
- Focal aware seizure: Patient is aware of the ictal symptoms during focal seizure activity. This is often seen with frontal, parietal, and lateral or non-dominant temporal seizures.
- An example is a patient who can report a “Jacksonian march” seizure semiology. This can be more formally identified as a focal motor aware seizure.
- Focal seizure with impaired awareness: Self-awareness is not maintained with focal seizure activity, often due to involvement of the hippocampus. An example is a patient who has a temporal lobe seizure with associated semiology of starring and unresponsiveness.
- Formerly called complex partial seizures.
- Expanded classification of focal seizures adds more details regarding ictal semiology; Motor Onset (automatism, clonic, hyperkinetic, etc.), or Non-Motor Onset (behavior arrest, sensory, emotional, etc.)
- History or video recording of ictal semiology can help localize the epileptogenic focus:
- Temporal lobe seizures:
- Temporal lobe epilepsy is the most common focal epilepsy.
- Mesial temporal seizures present with an aura of unusual smell, taste, automatisms (lip-smacking, hand rubbing), or feeling of déjà vu.
- Lateralized temporal lobe epilepsy presents with auditory auras.
- Can be seen secondary to an autosomal dominant mutation to the LGI1 gene.
- Amygdala-onset seizures present with fear and autonomic symptoms (tachycardia, piloerection).
- Parietal lobe seizures: Can present with hemibody paresthesias or other primarily sensory manifestations.
- Frontal lobe seizures: Diverse, but often involve hyperkinetic or bilateral atypical movements, such as clapping and leg bicycling. These may be brief and sudden, arise from sleep, and have minimal post-ictal confusion.
- Seizures of the supplementary motor area (SMA) of the frontal lobe present with fencer posturing:
- Extension of the contralateral arm, flexion of ipsilateral arm. Head and eyes will face the contralateral side.
- Seizures of the supplementary motor area (SMA) of the frontal lobe present with fencer posturing:
- Occipital lobe seizures: Can present with visual phenomena.
- Temporal lobe seizures:
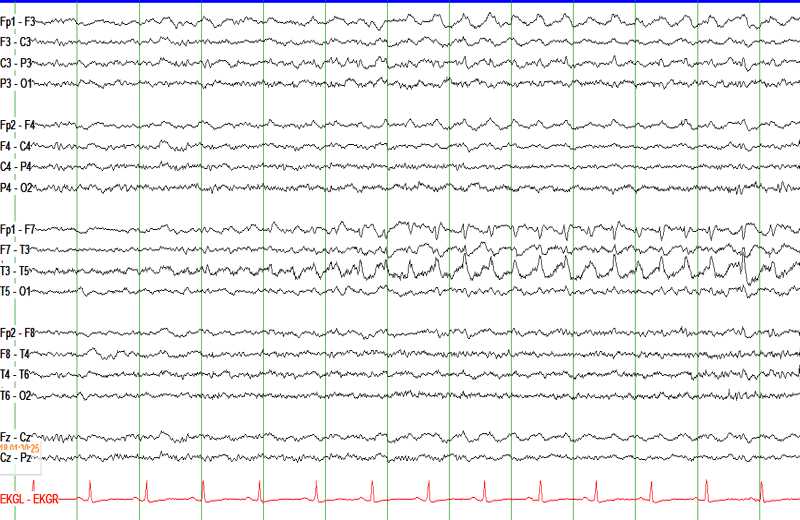
- Just because someone has seizures doesn’t mean they have epilepsy. For example, a patient with provoked seizures due to alcohol withdrawal does not have epilepsy. Seizures that are the result of an acute direct or indirect brain insult are termed “provoked” seizures.
- Most provoked seizures that occur secondary to an indirect brain insult (drug toxicity, electrolyte abnormality, etc.) present with generalized tonic-clonic seizures.