

Astrocytomas
- A slow-growing astrocytic tumor composed of bipolar “hair-like” (pilocytic) cells.
- Most common glioma in children.
- Associated with tuberous sclerosis, neurofibromatosis type 1 (NF1), and Li-Fraumeni syndrome.
- Optic nerve and chiasm glioma are associated with NF1.
- Often presents with symptoms of increased ICP (headache, nausea/vomiting), vision loss, ataxia, or cranial nerve deficits depending on location.
- Imaging: Cystic mass with a contrast rim-enhancing nidus or mural nodule with minimal vasogenic edema, dorsally exophytic. Most commonly found in the cerebellum.
- Also prefers midline structures such as the brainstem, optic chiasm, hypothalamus, and deep gray matter (basal ganglia).

Pilocytic Astrocytoma
- Also prefers midline structures such as the brainstem, optic chiasm, hypothalamus, and deep gray matter (basal ganglia).
- KIAA1549-BRAF gene fusion is characteristic of this tumor type.
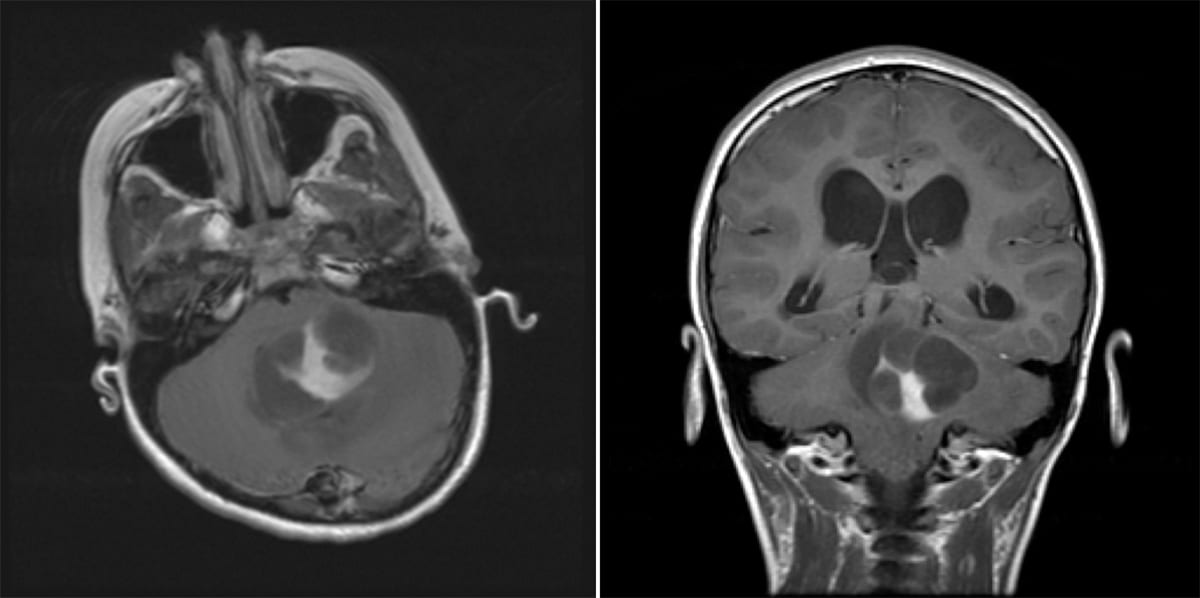
- 90% 10-year overall survival. It can be treated with surgical resection alone, and rarely progresses to malignant glioma.
- Classic patient presentation: Child presenting with increased ICP/ataxia, found to have a cerebellar cystic mass lesion with an enhancing mural nodule.
Subependymal giant cell astrocytoma (SGCA or SEGA)
- WHO grade 1 tumor almost exclusively seen in pediatric patients with tuberous sclerosis (TS) and before the age of 20.
- Seen in 5-15% of patients with TS.
- Often asymptomatic, but when symptomatic presents with obstructive hydrocephalus due to location in the foramen of Monro.
- Imaging: Well-circumscribed, partially-calcified intraventricular contrast-enhancing mass near the foramen of Monro.

Subependymal Giant Cell Astrocytoma
- WHO grade 1 tumor almost exclusively seen in pediatric patients with tuberous sclerosis (TS) and before the age of 20.
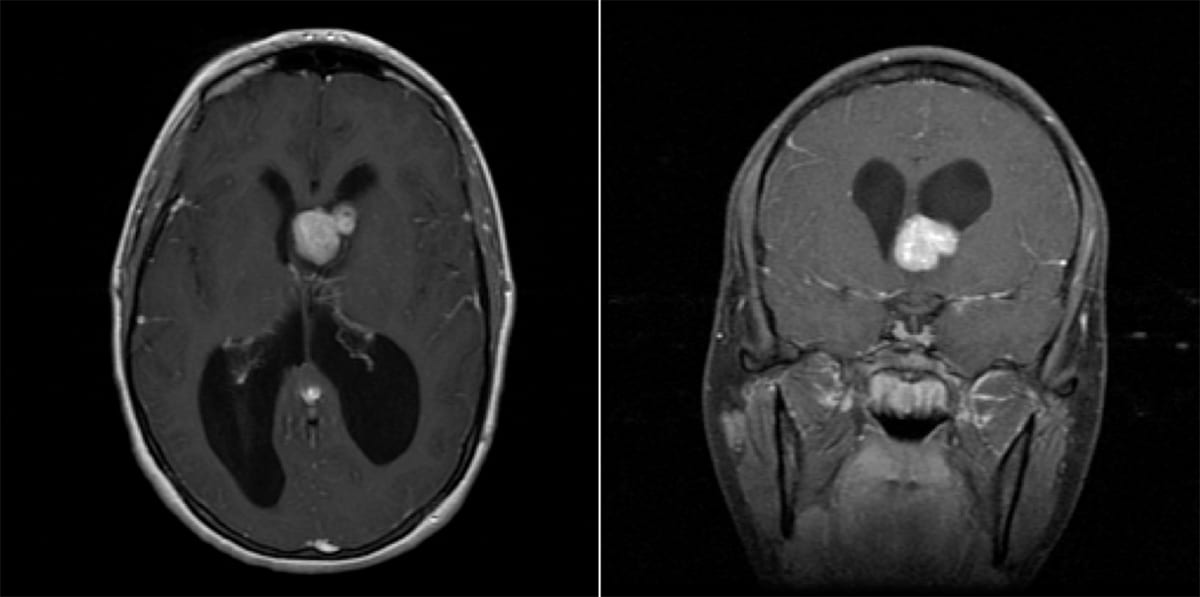
- Generally treated initially with mTOR inhibition with everolimus.
- If acute symptomatic or growing, can be treated with surgical resection.
- Tuberous Sclerosis Review: classically presents with seizures, mental retardation, and adenoma sebaceum. Associated with TSC2/tuberin (most cases) or TSC1/hamartin with cortical or subependymal tubers, hamartomas, renal angiomyolipomas, and cardiac rhabdomyomas.
- Generally treated initially with mTOR inhibition with everolimus.
Pleomorphic xanthoastrocytoma (PXA)
- Found in young patients who present with temporal lobe epilepsy.
- Imaging: Supratentorial peripheral cystic and contrast-enhancing mass abutting the leptomeninges with enhancing dural tail sign and scalloping of overlying bone.
- Associated with BRAFV600E mutations and homozygous CDKN2A/B deletions.
- Treated with surgical resection. However, local recurrence and malignant transformation are common so post-operative radiation is indicated for grade 3 tumors.
Adult-type diffuse gliomas
- Astrocytoma, IDH-mutant (grades 2-4)
- Patients present with progressive neurologic symptoms dependent on tumor location and/or with seizures.
- Imaging: T2-FLAIR mismatch sign is often present, with T2 hyperintensity and relative hypointensity on FLAIR sequences.
- MR Spectroscopy will have an elevated choline peak, low NAA peak, and elevated choline:creatinine ratio.
- Pathology:
- Grade 2: mitotic activity absent or low without microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor (homozygous deletion of CDKN2A/B).
- Grade 3: mitotic activity present without microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor.
- Formally known as anaplastic astrocytoma.
- Grade 4: microvascular proliferation, necrosis, or genetic markers that would upgrade the tumor present.
- Glioblastoma, IDH-wildtype (grade 4)
- Formally known as glioblastoma multiforme (GBM).
- The most common and also most destructive of the diffuse gliomas.
- Imaging: Contrast ring-enhancing lesions with significant vasogenic edema. Lesions can also have internal necrosis and can extend through the corpus callosum (butterfly lesion).
- Astrocytoma, IDH-mutant (grades 2-4)

Glioblastoma Multiforme
Left: Axial MRI, T2 FLAIR. Right: T1 w/ contrast. Note the hyperintensity on T1 w/ contrast.

Glioblastoma Multiforme
Left: Axial MRI, T2 sequence. Right: T2 w/ contrast. Note the central necrosis.
")
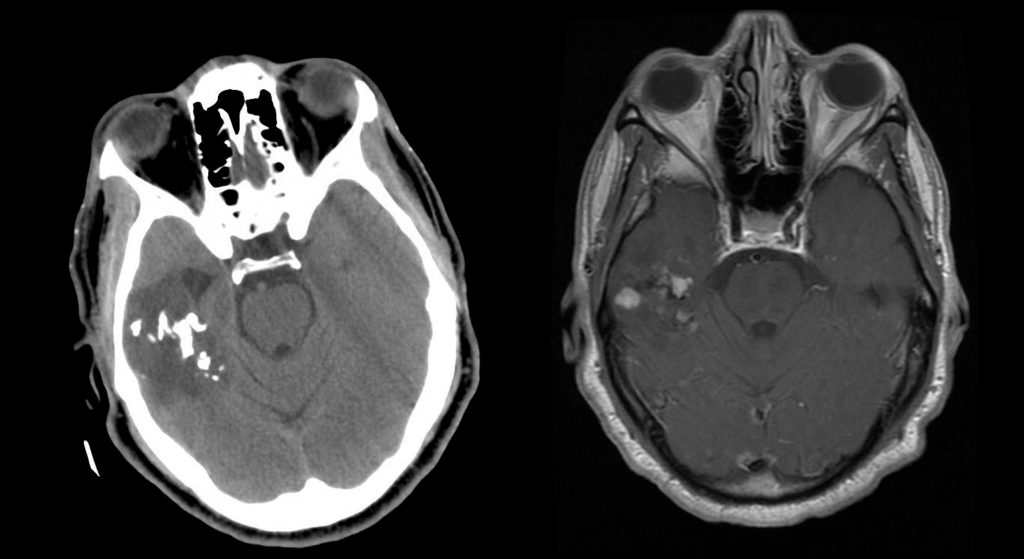
Glioblastoma Multiforme (GBM)
Left: Axial MRI, T1 w/ contrast. Middle: T2 FLAIR. Right: ADC.
- Genetics:
- IDH-1/2 mutations are associated with secondary GBM arising from a lower-grade glioma.
- Tumors without microvascular proliferation or necrosis can still be classified as “molecular” glioblastoma if genetic testing shows TERT promoter mutation, EGFR gene amplification, or gain of 7/loss of 10 chromosome copy number alterations.
- Pathology: Cells with increased mitotic activity with pseudopalisading necrosis and microvascular endothelial proliferation.
- Genetics:

Glioblastoma Multiforme
Low-power view showing palisading, or pseudo-palisading, necrosis with a rim of viable tumor-cell nuclei (arrows) lining up around a region of necrotic tumor cells (central pink region).

Glioblastoma Multiforme
Coronal gross cut. Large necrotic mass with increased vascularity and mass effect on the surrounding white matter.
- Treatment: Maximal safe resection followed by intensity-modulated radiation therapy (IMRT) plus concomitant temozolomide (alkylating chemotherapy) followed by adjuvant temozolomide.
- Methylation of the MGMT gene is associated with better treatment response to temozolomide.
- Bevacizumab, a monoclonal antibody that inhibits vascular endothelial growth factor (VEGF) can be used in recurrent or progressive GBM.
- Another alkylating agent, lomustine, is often used as a second-line treatment or in recurrent gliomas.
- Treatment: Maximal safe resection followed by intensity-modulated radiation therapy (IMRT) plus concomitant temozolomide (alkylating chemotherapy) followed by adjuvant temozolomide.
Oligodendrogliomas
- WHO grade 2 or WHO grade 3.
- Associated with 1p/19q co-deletion and IDH mutations;
- 1p/19q co-deletion patients have a better overall prognosis compared to astrocytic tumors which are 1p/19q normal.
- Imaging: Partially calcified T2 heterogeneous, hyperintense subcortical/cortical mass with patchy or minimal contrast enhancement. Most often found cortically in the frontal or temporal lobes.
Attributed by RadsWiki, CC BY-SA 3.0, via Wikimedia Commons, https://upload.wikimedia.org/wikipedia/commons/2/28/Oligodendroglioma_001.jpg, https://upload.wikimedia.org/wikipedia/commons/9/9b/Oligodendroglioma_007.jpg
- Commonly occurs in the 4th or 5th decade of life.
- Pathology: Cells with a “fried egg” appearance with monotonous round nuclei, surrounded by prominent perinuclear halos.
- Treatment: Surgical resection followed by radiation and chemotherapy.

Oligodendroglioma
Neoplastic oligodendrocytes with rounded nuclei, lacking abundant fibrillary processes, and with perinuclear halos giving it a “fried egg” appearance.

Oligodendroglioma
Neoplastic oligodendrocytes with rounded nuclei, lacking abundant fibrillary processes, and with perinuclear halos giving it a “fried egg” appearance.